Что такое заболевание ксеродерма
Заболевание развивается обычно у новорожденных или в раннем детском возрасте, редко, в основном при пигментном ксеродермоиде, — на втором десятилетии жизни.Распространенность в популяции европеоидной расы составляет 2, 3 случая на 1 млн новорожденных, общая — 4:1 000 000. Тип наследования аутосомно-рецессивный.
Заболевание генетически неоднородное. В зависимости от степени и характера нарушений репарации ДНК выделяют 7 групп комплементации (от A до G) и «вариант пигментной ксеродермы», или пигментный ксеродермоид. Cиндром де Санктиса–Каккьоне может быть при любом типе заболевания, но чаще его выявляют в группе коплементации А. Мутантные гены расположены на следующих хромосомах: 9q22.3 (тип A), 2q21 (тип B), 3p25 (тип C), 19q13.2 (тип D), 11p12 (тип E), 16p13.3 (тип F), 13q13 (тип G), 6p21.1-p12 (пигментный ксеродермоид), 10q11 (синдром де Санктиса-Каккьоне).
В основе развития заболевания лежит генетическое нарушение репликативного и репаративного синтеза ДНК в результате дефекта ее ферментов — полимеразы и эндонуклеазы, приводящее к разрушению ДНК коротковолновым спектром УФЛ-UVB (в пределах волн 280-310 нм), что ведет к несвоевременному и слабому накоплению тимидина в эпидермоцитах, фибробластах и лимфоцитах.
При рождении кожные покровы чистые. Первые симптомы развиваются в возрасте 6 мес или несколько позже.Прогноз при пигментной ксерордерме плохой вследствие развития злокачественных опухолей с быстрым прогрессирующим течением, метастазами и летальным исходом у многих больных в возрасте 10-15 лет. Риск развития злокачественных опухолей кожи повышен в сотни раз по сравнению с общей популяцией.
При отсутствии защиты от солнца базальноклеточный или плоскоклеточный рак обычно развивается в возрасте 8–9 лет.Значительно повышен риск развития меланомы, а также новообразований внутренних органов (легких, молочной железы, поджелудочной железы, желудка, головного мозга) и лейкоза.
В течении пигментной ксеродермы условно выделяют пять стадий:
Эритематозная стадия
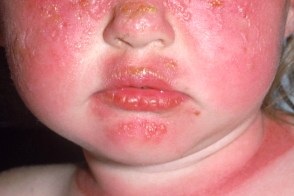 Развивается уже на первом году жизни. После непродолжительной инсоляции на открытых участках появляются эритематозно-сквамозные отечных пятен, часто с везикулами или пузырями. Развивается уже на первом году жизни. После непродолжительной инсоляции на открытых участках появляются эритематозно-сквамозные отечных пятен, часто с везикулами или пузырями. |
Стадия гиперпигментации
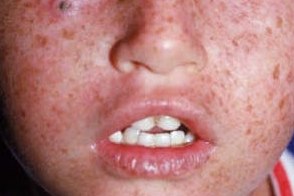 Характеризуется образованием множественных пигментных пятен типа веснушек и лентиго, разных по форме, размеру и интенсивности окраски (от светло- до темно-коричневой). Характеризуется образованием множественных пигментных пятен типа веснушек и лентиго, разных по форме, размеру и интенсивности окраски (от светло- до темно-коричневой). |
Стадия атрофии
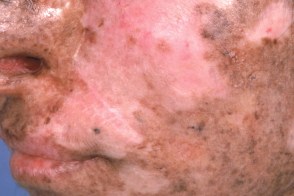 При этой стадии кожа истончается, становится сухой, напряженной, появляются гладкие атрофические рубцы с телеангиэктазиями, гипо- и/или гиперпигментацией (пойкилодермия). Атрофические изменения с очагами склероза приводят к формированию микростомии, эктропиона, сужению отверстий носа, истончению ушной раковины и кончика носа. При этой стадии кожа истончается, становится сухой, напряженной, появляются гладкие атрофические рубцы с телеангиэктазиями, гипо- и/или гиперпигментацией (пойкилодермия). Атрофические изменения с очагами склероза приводят к формированию микростомии, эктропиона, сужению отверстий носа, истончению ушной раковины и кончика носа. |
Стадия доброкачественных опухолей
Характеризуется возникновением ангиом, фибром, папиллом, кератоакантом, актинического кератоза.Может наблюдаться уже в детском возрасте (2–5 лет)
Стадия малигнизации
Наступает через 8-10 лет от начала заболевания в виде озлокачествления предшествовующих доброкачественных новообразований либо самостоятельно формирующихся меланомы, саркомы, плоскоклеточной и базальноклеточной карциномы.Иногда злокачественные опухоли появляются очень рано, уже в первые годы жизни.
Поражение глаз
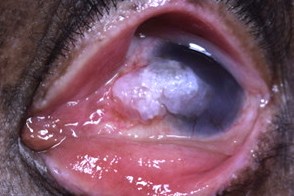 Патология глаз встречается у 40% больных.У большинства больных отмечается светобоязнь (она может быть с самого начала болезни), конъюнктивит (может быть самым ранним проявлением), кератит, блефарит, гиперпигментация радужки, телеангиэктазии и пигментация век и конъюнктивы, снижение остроты зрения. Позднее возможно развитие симблефарона, помутнения роговицы, рост доброкачественных и злокачественных опухолей конъюнктивы. Патология глаз встречается у 40% больных.У большинства больных отмечается светобоязнь (она может быть с самого начала болезни), конъюнктивит (может быть самым ранним проявлением), кератит, блефарит, гиперпигментация радужки, телеангиэктазии и пигментация век и конъюнктивы, снижение остроты зрения. Позднее возможно развитие симблефарона, помутнения роговицы, рост доброкачественных и злокачественных опухолей конъюнктивы. |
Другие нарушения
Возможны эндокринные нарушения; неврологические расстройства; иммунодефицит со стойкой анемией и большим количеством бластных клеток в крови.
Синдром де Санктиса-Каккьоне
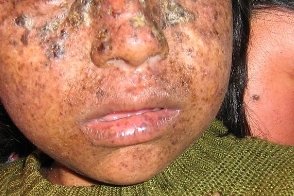 Известен также как de Sanctis-Cacchione syndrome и идиотия ксеродермическая. Известен также как de Sanctis-Cacchione syndrome и идиотия ксеродермическая.Отличается особо тяжелым течением. Характеризуется ранним началом тяжелых кожных проявлений, грубыми нарушениями со стороны ЦНС (микроцефалия, идиотия, недоразвитие мозжечка, потеря слуха и др.), гипогонадизмом, карликовым ростом, укорочением ахиллова сухожилия. Рентгенологически выявляют уменьшение турецкого седла, могут быть нарушения порфиринового обмена. |
Диагноз пигментной ксеродермы устанавливают на основании характерной клинической картины и данных гистологического исследования. Возможна пренатальная диагностика с выявлением мутантных генов.
- Синдром Блума
- Синдром Коккейна
- Наследственная эритропоэтическая порфирия
Лечение не разработано. Необходимо строго ограничивать пребывание на солнце, использовать физические и химические фотозащитные средства в течение всего дня, антиоксиданты, препараты интерферона. Большое значение придают раннему удалению опухолей, назначению с профилактической целью препаратов витамина А, системных ретиноидов.
Для лечения актинического кератоза и ранних стадий немеланозного рака кожи применяют имиквимод, фторурацил и фотодинамическую терапию.С целью раннего выявления опухолей проводят профилактический осмотр больных каждые 3 мес. Лечение новообразований осуществляют стандартными методами.
Что такое заболевание ксеродерма
Этиология и встречаемость пигментной ксеродермы. Пигментная ксеродерма — генетически разнородное, панэтническое, аутосомно-рецессивное заболевание репарации ДНК, вызывающее выраженную гиперчувствительность к ультрафилетовым лучам. В США и Европе распространенность составляет приблизительно 1 на миллион, но в Японии — 1 на 100 000.
Патогенез пигментной ксеродермы
Репарация ДНК, повреждаемой ультрафиолетовым излучением, происходит благодаря трем механизмам: эксцизионной репарации, пострепликационной репарации и фотореактивизации. Эксцизионная репарация устраняет повреждения ДНК восстановлением нуклеотида или одного основания. Пострепликационная репарация — аварийный механизм, обеспечивающий репликацию ДНК вдоль поврежденного шаблона. Фотореактивация возвращает поврежденную ДНК в нормальное химическое состояние без удаления или замены генетического материала.
Нуклеотидная эксцизионная репарация — сложный разносторонний процесс, в котором участвуют, по крайней мере, 30 белков. Основной принцип — удаление небольшого однонитевого участка ДНК, содержащего повреждение, разрезами с обеих сторон поврежденного сегмента, с последующим заполнением промежутка вновь синтезированной нитью на основе неповрежденной дополнительной нити, используемой как шаблон. В ходе транскрипции гена повреждение ДНК блокирует продвижение РНК-полимеразы II.
Остановленная РНК-полимераза II инициирует эксцизионную репарацию нуклеотидов (репарация, вызванная транскрипцией). В остальной части генома и в нетранскрибируемых генах комплекс нуклеотидной эксцизионной репарации выявляет повреждения ДНК по искажению скручивания ДНК (глобальная геномная репарация).
Иногда нуклеотидная эксцизионная репарация неспособна исправить повреждение ДНК до репликации. Поскольку такие повреждения тормозят репликацию ДНК, пострепликационная репарация обходит повреждение, позволяя продолжить синтез ДНК. ДНК-полимераза n обеспечивает ДНК синтез через поврежденные участки; она эффективно и точно катализирует правильный синтез, пропуская дитимидиновые повреждения.
Пигментная ксеродерма вызвана мутациями, влияющими на геномную нуклеотидную эксцизионную репарацию или пострепликационную репарацию. В отличие от нее, синдром Коккейна — сходное заболевание, вызываемое мутациями, влияющими на связанный с транскрипцией путь нуклеотидной эксцизионной репарации. И пигментная ксеродерма, и синдром Коккейна подразделяют на 10 биохимических групп комплементации; каждая группа отражает мутации различных компонентов нуклеотидной эксцизионной или пострепликационной репарации.
Снижение или полное отсутствие возможности глобальной геномной репарации или пострепликационной репарации приводит к потере функций, необходимых для поддержания целостности генома, и вызывает накопление онкогенных мутаций. Новообразования у пациентов с пигментной ксеродермой имеют более высокий уровень мутаций генов-онкогенов и супрессоров опухолевого роста, чем опухоли в нормальной популяции, и эти мутации специфично вызывает ультрафиолетовое облучение.
Фенотип и развитие пигментной ксеродермы
Симптоматика у пациентов с пигментной ксеродермой развивается в среднем в возрасте 1-2 года, хотя приблизительно у 5% пациентов описано начало после 14 лет. Первые симптомы обычно включают легкие ожоги, повышенную фоточувствительность, появление лентигинозной сыпи и фотобоязнь. Постоянное воздействие вызывает преждевременное старение кожи (истончение, морщины, веснушки, телеангиэктазии), фотокератоз и доброкачественные и злокачественные новообразования. Почти у 45% пациентов развиваются базальноклеточные или сквамозноклеточные карциномы, у 5% — меланомы.
Приблизительно 90% карцином появляется в местах сильного ультрафиолетового воздействия — на лице, шее, голове, кончике языка. До введения профилактических мероприятий средний возраст развития новообразований был 8 лет, на 50 лет раньше, чем в общей популяции, а их частота более чем в 1000 раз выше общепопуляционной.
Кроме кожных симптомов, у 60-90% пациентов отмечают нарушения со стороны глаз, включая фотобоязнь, конъюнктивиты, блефариты, эктропион и новообразования. Распределение патологии глаз и новообразований соответствуют участкам наибольшего влияния ультрафиолета.
Приблизительно у 18% пациентов развивается прогрессирующая дегенерация нейронов. Симптоматика представлена нейросенсорной глухотой, задержкой умственного развития, спастичностью, гипорефлексиией или арефлексией, сегментарной демиелинизацией, атаксиями, хореоатетозом и надъядерной офтальмоплегией. Тяжесть неврологических симптомов обычно пропорциональна степени нарушения нуклеотидной эксцизионной репарации. Нейродегенерация может быть вызвана неспособностью исправлять ДНК, повреждаемую эндогенными свободными радикалами.
Эксцизионная нуклеотидная репарация также исправляет повреждения ДНК, вызванные большинством химических канцерогенов, например сигаретным дымом, подгоревшей пищей или цисплатином (производное платины с противоопухолевой активностью, способное стимулировать механизмы клеточно-опосредованной цитотоксичности). В связи с этим у пациентов в 10-20 раз повышена частота опухолей внутренних органов, например опухолей мозга, лейкозов, рака легких и желудка.
Пациенты с пигментной ксеродермой имеют уменьшенный срок жизни; без профилактической защиты срок их жизни почти на 30 лет короче по сравнению с лицами без пигментной ксеродермы. Наиболее частые причины смерти — метастатические меланомы и сквамозноклеточные карциномы кожи.
Два сходных с пигментной ксеродермой заболевания, синдром Коккейна и трихотиодистрофия, также вызываются дефектами в различных компонентах клеточного механизма репарации нарушений ДНК, вызванных ультрафиолетом. Для обоих заболеваний характерны плохой постнатальный рост, уменьшение подкожной клетчатки, контрактуры суставов, тонкая кожа с фоточувствительностью, умственная задержка и неврологические симптомы.
У детей с синдромом Коккейна также развиваются дегенерация сетчатки и глухота; дети с трихотиодистрофией имеют ихтиоз и ломкие волосы и ногти. При обоих синдромах пациенты редко живут дольше 20 лет. Интересно, что ни тот, ни другой синдром не сопровождаются увеличением частоты опухолей кожи. Тем не менее дефекты в некоторых генах репарации ДНК (ERCC2, ERCC3 и ERCC5) приводят к фенотипам, имеющим общие симптомы между пигментной ксеродермой, синдромом Коккейна и трихотиодистрофией.
Особенности фенотипических проявлений пигментной ксеродермы:
• Возраст начала: детство
• Чувствительность к ультрафиолету
• Опухоли кожи
• Неврологическая дисфункция
Лечение пигментной ксеродермы
Подтверждение диагноза пигментной ксеродермы проводят функциональными тестами репарации ДНК и чувствительности к ультрафиолету; такие тесты обычно выполняют на зрелых фибробластах кожи. Подтверждение диагноза идентификацией мутаций в генах, связанных с пигментной ксеродермой, в настоящее время клинически недоступно.
Оказание помощи пациентам с пигментной ксеродермой состоит в исключении воздействия солнечного света, ношении защитной одежды, использовании физических и химических солнцезащитных кремов и тщательном наблюдении для своевременного выявления и удаления злокачественных новообразований. Исцеляющее лечение в настоящее время недоступно.
Риски наследования пигментной ксеродермы
Поскольку пигментная ксеродерма — аутосомно-рецессивное заболевание, семейный анамнез у большинства пациентов не отягощен. Для родителей, имеющих ребенка с пигментной ксеродермой, риск для последующих детей — 1 к 4. Пренатальная диагностика возможна путем определения функции репарации ДНК и чувствительности к ультрафиолету в культуре амниоцитов или клеток ворсин хориона.
Пример пигментной ксеродермы. B.C., 3-летний мальчик, направлен в клинику дерматологии в связи с выраженной чувствительностью к солнечным лучам и появлением множественных пятен. При клиническом осмотре выявлены фотобоязнь, конъюнктивит и значительная лентигинозная гиперпигментация в незащищенных от солнца областях; в остальном его развитие и данные осмотра оказались нормальными. Пациент — ребенок от неродственного брака родителей японского происхождения; никто в семье подобным образом не болел. Дерматолог сказал, что у ребенка классические проявления пигментной ксеродермы.
Для подтверждения диагноза проведена биопсия кожи с целью оценить репарацию ДНК и чувствительность к ультрафиолетовому излучению фибробластов кожи. Результаты этого теста подтвердили диагноз пигментной ксеродермы. Несмотря на соответствующие профилактические меры, в возрасте 15 лет у ребенка развилась метастатическая меланома, и через 2 года он умер. У его родителей есть два других ребенка; ни один из них не болен пигментной ксеродермой.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Пигментная ксеродерма
Пигментная ксеродерма — одна из наследственных дерматологических болезней. Встречается она крайне редко: 2 случая на 1 миллион новорожденных. Симптоматика заболевания проявляется уже в раннем детском возрасте и неуклонно прогрессирует без комплексного лечения. Опасность пигментной ксеродермы заключается в ее высоком онкогенном потенциале. Кожные проявления болезни являются облигатным предраком, то есть склонны к злокачественному перерождению и практически во всех случаях переходят в рак.
Вместе с тем, первые дерматологические симптомы пигментной ксеродермы у ребенка чаще всего расцениваются родителями, как явления какого-либо аллергического дерматита и не вызывают излишнего беспокойства. Отсутствие настороженности приводит к позднему обращению за медицинской помощью и запоздалому лечению.
Причины и механизм развития
Пигментная ксеродерма относится к группе наследственных заболеваний и передается аутосомно-рецессивным путем. Под аутосомно-рецессивным понимают наследование, когда мутации происходят в любых хромосомах, кроме половых, и для проявления заболевания у ребенка необходимо, чтобы у обоих супругов был дефектный ген.
Основные звенья патогенеза пигментной ксеродермы — повышенная чувствительность к прямым солнечным лучам и невозможность организма устранять повреждения ДНК, вызванные ультрафиолетовым излучением. Нарушение процессов восстановления ДНК связано с отсутствием или низкой активностью в коже ферментов, отвечающих за репарацию: нуклеозидов и протеаз.
Необратимые повреждения генетического аппарата клеток приводят к ряду патологических процессов. Так, на ранней стадии заболевания в кожных покровах наблюдается:
- усиление скорости деления рогового слоя кожи и слущивание отмерших чешуек;
- истончение шиповатого (промежуточного) слоя;
- избыточное отложение меланина в базальном (ростковом) слое эпидермиса и в сосочковом слое дермы;
- инфильтрация лейкоцитами и другими клетками воспаления околососудистых пространств.
На более поздних стадиях пигментной ксеродермы на коже обнаруживают участки избыточного ороговения (гиперкератоз), акантоза (гиперпролиферативные очаги, нередко пигментированные) с элементами клеточной атипии, дегенеративные изменения в коллагеновых и эластиновых волокнах.
Терминальные стадии заболевания преимущественно характеризуются атрофией (истончением) и изъязвлением участков кожи, реже встречаются бородавчатые или папилломатозные разрастания. Особенно выражены эти процессы на открытых частях тела.
В зависимости от повреждённой хромосомы, выделяют 8 подвидов заболевания: A, B, C, D, E, F G и пигментный ксеродермоид Юнга. Первые 7 типов пигментной ксеродермы имеют схожее клиническое течение и определяются лишь после молекулярно-генетического анализа. Пигментный ксеродермоид Юнга имеет более благоприятный прогноз — симптоматика заболевания появляется позже, а сам патологический процесс имеет легкое течение.
Самостоятельной клинической формой заболевания является синдром Де Санктиса-Каккьоне. Он считается самой агрессивной разновидностью пигментной ксеродермы и характеризуется выраженными изменениями со стороны центральной нервной системы. Синдром может развиться при любом хромосомном варианте заболевания, но чаще всего встречается при подтипе A.
Клиническая картина
У новорожденных детей симптомов заболевания нет. Первые признаки пигментной ксеродермы начинают замечать в возрасте от 3 месяцев до 4 лет. В казуистических случаях дебют заболевания возможен в зрелом возрасте. В литературе имеются данные о появлении симптоматики у пациентов 25-30 лет.
Пигментная ксеродерма характеризуется сезонностью и стадийностью течения. Обострение заболевания происходит в солнечное время года (весна — ранняя осень). Первая стадия называется воспалительной и проявляется специфической триадой симптомов:
- фоточувствительность — непереносимость прямых лучей солнечного света;
- эритема — покраснение кожных покровов открытых участков тела после пребывания на солнце;
- очаги гиперпигментации — появление участков избыточной окраски.
Эритема кожных покровов носит стойкий характер. Она нередко сопровождается выраженной отечностью, шелушением и образованием небольших пузырьков с прозрачным содержимым. После стихания воспалительных процессов на месте эритемы появляются очаги гиперпигментации в виде светло- или темно-коричневых пятен разных размеров. В большинстве случаев они напоминают россыпь веснушек, но могут иметь большие размеры (лентиго).
Вторая стадия — пойкилодермическая. Она начинается в возрасте 3-7 лет и характеризуется выраженными изменениями кожных покровов в виде телеангиоэктазий (сосудистых «звездочек»), участков гиперкератоза с обильным шелушением, очагов атрофии и истончения кожи. Типичным проявлением второй стадии также является образование небольших гладких рубчиков белесоватого цвета с блестящей поверхностью. При внимательном осмотре участки атрофии кожи имеют неровный шероховатый вид и похожи на ожоговую поверхность.
Выраженные изменения кожных покровов приводят к ряду серьезных последствий:
- деформация наружного носа и носовых ходов, как следствие — затруднение дыхания;
- искривление рта;
- деформация ушей за счет атрофии хрящей;
- выпадение ресниц;
- выворот или заворот век.
У пациентов с пигментной ксеродермой сохраняются очаги гиперпигментации, а с прогрессированием заболевания появляются гипопигментированные пятна. Как правило, они локализуются на спинке и крыльях носа, на подбородке.
У 75-80% пациентов на второй стадии также определяются признаки поражения органа зрения: слезотечение и светобоязнь, блефарит, изъязвление конъюнктивы и ее воспаление, помутнение роговицы и появление на ней пятен. У 20% больных выявляются симптомы нарушения работы центральной нервной системы в виде снижения чувствительности, умственной отсталости, изменения согласованной работы мышц (атаксия) и снижения или отсутствия безусловных рефлексов (например, сухожильных).
В третью (опухолевидную) стадию заболевание переходит в подростковом возрасте. Для нее характерно появление на пораженных очагах доброкачественных или злокачественных новообразований различной формы и величины. Доброкачественные опухоли могут быть представлены папилломами, невусами, фибромами, ангиомами и имеют высокий риск онкологической трансформации. Нередко новообразования также выявляют на слизистой оболочке полости рта и носа.
Тяжелое клиническое течение имеет синдром Де Санктиса-Каккьоне. Его основные симптомы — нарушение умственного развития (идиотия) из-за уменьшения размеров головного мозга и черепа, карликовость и типичные кожные проявления пигментной ксеродермы. Дополнительными признаками синдрома является задержка полового развития и тяжелые нарушения работы нервной системы (парезы, параличи, атаксия). Кожные проявления у пациентов с синдромом Де Санктиса-Каккьоне появляются рано. Они ярко выражены, быстро прогрессируют и переходят в онкологические новообразования уже в детском возрасте.
Осложнения
Основное осложнение заболевания — появление кожных и висцеральных (внутренних) злокачественных опухолей. Наиболее высоким риском онкогенной трансформации обладают множественные бородавчатые новообразования. Частота возникновения злокачественных опухолей также коррелирует с тяжестью кожных патологических процессов. При раннем начале заболевания и его агрессивном течении онкологические новообразования появляются в подростковом возрасте (12-14 лет) и быстро метастазируют во внутренние органы.
Среди злокачественных опухолей чаще всего развивается плоско- или базальноклеточный рак, реже всего — меланома кожи или глаза. Описаны случаи возникновения более 50 различных вариантов онкологии: фибросарком, ангиосарком, гистиоцитом и других.
Заболевание также может приводить к выраженным деформациям лица (рта, носа, ушей). Как результат — социальная дезадаптация ребенка и психологические проблемы.
Диагностика
Большое значение в постановке правильного диагноза имеет детальный сбор анамнеза. Собранные данные (начало болезни в раннем детском возрасте, сезонность симптоматики, высокая фоточувствительность, кровные браки) позволяют предположить пигментную ксеродерму. Диагноз подтверждается после цитологического и гистологического исследования пораженных участков кожи.
Среди дополнительных обследований — осмотр стоматолога, оториноларинголога, офтальмолога и невролога для исключения поражения центральной нервной системы, ротового аппарата, органов зрения и слуха. При синдроме Де Санктиса-Каккьоне также проводится рентгенологическое исследование или компьютерная томография костей черепа, магнитно-резонансная томография головного мозга. При этом обнаруживают малые размеры турецкого седла, уменьшение объема черепной коробки и головного мозга, недоразвития мозжечка и гипофиза.
Методы лечения
Этиопатогенетического лечения пигментной ксеродермы не существует. По своей сущности, любые способы терапии являются симптоматическими и направлены на устранение имеющихся проявлений заболевания. В комплексное лечение заболевания входят:
- курсы витаминотерапии;
- противомалярийные препараты — отмечена эффективность лекарств этой группы в устранении воспалительного процесса и сенсибилизации организма;
- ароматические ретиноиды;
- цитостатические противоопухолевые препараты.
Пациентам также рекомендуется скорректировать образ жизни, максимально ограничить пребывание на солнце, а перед выходом на улицу позаботиться о защите кожных покровов от солнечных лучей.
Все доброкачественные кожные элементы подлежат обязательному удалению и гистологическому исследованию. При трансформации новообразования в злокачественное тактика лечения определяется, исходя из типа опухоли и стадии онкологического процесса.
Прогноз
Качество и продолжительность жизни зависят от нескольких факторов, в первую очередь, от сроков обращения за медицинской помощью, постановки правильного диагноза и раннего начала комплексного лечения. При своевременной диагностике заболевания и постоянном динамическом наблюдении врачом-дерматологом или онкодерматологом прогноз относительно благоприятный.
При бородавчатых разрастаниях на коже, около 35% пациентов не доживают до совершеннолетия и погибают в возрасте 13-15 лет. Это связано с высокой тенденцией к озлокачествлению и быстрым метастазированием опухоли во внутренние органы.
При синдроме Де Санктиса-Каккьоне прогноз неблагоприятный. Агрессивное течение заболевания приводит к летальному исходу от онкопатологии еще в детском возрасте у 70% пациентов.
Пигментная ксеродерма: что это за болезнь?

Пигментная ксеродерма – это болезнь, также известная под названием лентикулярный меланоз или злокачественное лентиго. Это достаточно редкая хроническая патология: она наблюдается у одного человека из 260 тысяч населения и заключается в чрезмерной чувствительности кожи к ультрафиолету. Предполагается, что эта патологическая реакция – часть легенды о воздействии солнечного света на «вампиров» наряду с порфирией. На сегодняшний день данная болезнь не поддается излечению, врачи могут предложить больным с таким диагнозом только придерживаться особенного образа жизни. Медикаментозное лечение является симптоматическим и не дает стойкого положительного эффекта.
Причины развития пигментной ксеродермы
Ученые открыли два различных генотипа лентикулярного меланоза.
Один из них может передаваться по аутосомно-доминантному пути, а второй — по аутосомно-рецессивному. Развитие болезни происходит по причине недостаточности определенных ферментов, несущих ответственность за полноценное восстановление ДНК после ее травматизации лучами ультрафиолета. Из-за такой особенности больные пигментной ксеродермой после нахождения в условиях избыточной солнечной радиации страдают от повреждений кожных покровов и глаз. Иногда болезнь протекает в скрытой форме, в такой ситуации она вызывает лишь пигментные нарушения.
Пигментная ксеродерма чаще встречается среди жителей Среднего Востока, а также средиземноморского побережья африканского континента. Заболевание может возникать у людей разного пола, однако некоторые исследования показывают, что риск его развития у девочек несколько выше.
Для данной болезни характерна повышенная вероятность предрасположенности к ксеродерме при близкородственных браках, что подтверждает исследование 2016 года (American Journal of Human Biology) о частоте возникновения тяжелых аутосомно-рецессивных заболеваний у детей.
Признаки болезни

Первые проявления пигментной ксеродермы обычно возникают у детей раннего возраста — до двух-трех лет жизни. Второй по распространенности период начала заболевания – 20-45 лет.
Болезнь манифестирует в летнее время года во время пиковой активности солнечных лучей. Крайне редки случаи, когда заболевание впервые проявлялось у подростков или пожилых людей, однако такие ситуации тоже известны.
Для пигментной ксеродермы характерны пять последовательных стадий развития.
- Эритематозная стадия
На раннем этапе своего возникновения болезнь проявляется симптоматикой фотодерматита, появлением красных пятен, сыпи и отечности после нахождения на солнце. В редких случаях могут возникать везикулы на пораженных участках кожи.
- Стадия гиперпигментации
При дальнейшем прогрессировании болезни красноватые пятна на коже превращаются в участки стойкой пигментации. Новые отметины могут иметь различный оттенок — от светло-коричневатого до практически черного.
- Атрофия пораженных участков
Пигментированная кожа в дальнейшем начинает истончаться, пересыхать и атрофироваться, что объясняется течением воспалительных процессов. Подобное развитие событий чревато серьезными изменениями физиологических отверстий — уменьшением рта по контуру, заращением ноздрей, истончением ушных раковин или крыльев носа. Практически всегда болезнь сопровождается воспалительным поражением конъюнктивы (конъюнктивитом).
- Гиперкератическая стадия
При дальнейшем прогрессировании кожа на тех областях, которые подвергаются воздействию ультрафиолетовых лучей, покрывается твердыми узелками, папилломами, бородавками и доброкачественными опухолевыми образованиями. Со временем такие опухоли склонны к перерождению в злокачественные.
Это состояние считают последней стадией пигментной ксеродермы. Онкологическое заболевание может развиться примерно спустя десять лет после проявления первых симптомов болезни. Но случается и так, что злокачественные опухоли образуются гораздо раньше. Онкологическое поражение при пигментной ксеродерме может иметь вид меланомы, саркомы, базалиомы.
На сегодняшний день пигментная ксеродерма рано или поздно приводит к раку. Средний возраст смертности при выявлении ксеродермы в раннем детстве – 16 лет.
Болезнь склонна к постоянному прогрессированию с возрастом, ее симптомы появляются даже после краткосрочного нахождения под солнечными лучами. Дети с таким диагнозом также часто страдают от дистрофии костной системы и могут иметь некоторые проблемы с когнитивным и психическим развитием вследствие нарушений обмена веществ, дефицита витамина D и особенностей образа жизни, диктуемых заболеванием.
Терапия при ксеродерме

Пигментная ксеродерма требует постоянной профилактики осложнений заболевания и мер по облегчению состояния. К ним относят местное нанесение глюкокортикостероидов, препаратов-цитостатиков, витаминно-минеральные комплексы, увлажнение кожи, обеззараживание и ранозаживляющие составы при наличии повреждений кожных покровов из-за повышенной опасности инфицирования. Определенную эффективность показывают противомалярийные лекарственные средства.
Для составления рациона питания рекомендовано обращение к диетологу для восполнения дефицита витаминов, особенно в раннем детском возрасте.
При наличии кожных новообразований вирусной (бородавки) или иной природы показано срочное удаление.
К мерам профилактики относят также избегание солнечного света и ультрафиолетовых лучей как на улице, так и в помещении, использование кремов с высоким SPF (Sun Protective Factor, показатель защиты от солнца).
Специфическая терапия не разработана, хотя есть определенные обнадеживающие результаты исследований эффективности иммунотерапии для лечения онкологических заболеваний. Хотя результатов по лечению пигментной ксеродермы пока нет, есть вероятность, что данные методики будут также эффективны в борьбе со злокачественными новообразованиями, ассоциированными с ксеродермой.
ПИГМЕНТНАЯ КСЕРОДЕРМА: КЛИНИКО-ГЕНЕТИЧЕСКИЕ ОСОБЕННОСТИ И ТЕРАПЕВТИЧЕСКИЕ ПОДХОДЫ Текст научной статьи по специальности «Клиническая медицина»
Аннотация научной статьи по клинической медицине, автор научной работы — Белышева Т.С., Наседкина Т.В., Клецкая И.С., Волкова А.С., Семенова В.В.
Пигментная ксеродерма — редкое генетическое заболевание, характеризующееся повышенной чувствительностью кожи к повреждающему действию ультрафиолетового (УФ) излучения. У большинства заболевших (до 75%) первые симптомы возникают в раннем возрасте. Характерно хроническое солнечное поражение кожи со стадийностью изменений и риском развития злокачественных опухолей, величина которого зависит от участвующего гена. Помимо изолированных кожных проявлений, описаны неврологические нарушения, включающие прогрессирующие когнитивные дисфункции, нейросенсорную тугоухость, атаксию, пирамидные и экстрапирамидные расстройства, арефлексию. Лечение больных с пигментной ксеродермой преимущественно симптоматическое и превентивное (защита от УФ-излучения). В настоящее время ведутся разработки таргетных лекарственных препаратов для репарации ДНК, повышения устойчивости клеток к воздействию УФ-излучения и профилактики таким образом возникновения онкологических заболеваний.
Похожие темы научных работ по клинической медицине , автор научной работы — Белышева Т.С., Наседкина Т.В., Клецкая И.С., Волкова А.С., Семенова В.В.
XERODERMA PIGMENTOSUM: CLINICAL AND GENETIC FEATURES AND THERAPEUTIC APPROACHES
Xeroderma pigmentosum is rare genetic disorder characterized by increased skin sensitivity to damaging ultraviolet (UV) light. First symptoms manifest at early age in most cases (up to 75%). Chronic damage due to sun exposure is common, it has different stages of changes and risk of further development of malignant tumors that depends on the gene involved. Additionally to skin manifestations there are various neurological disorders such as progressive cognitive dysfunctions, sensorineural hearing loss, ataxia, pyramid and extrapyramidal disorders, areflexia. Treatment of patients with xeroderma pigmentosum is mostly symptomatic and preventive (protection against UV). Nowadays targeted medications for DNA repair and increasing cells resistance to UV light, thus preventing the oncological diseases, are under development.
Текст научной работы на тему «ПИГМЕНТНАЯ КСЕРОДЕРМА: КЛИНИКО-ГЕНЕТИЧЕСКИЕ ОСОБЕННОСТИ И ТЕРАПЕВТИЧЕСКИЕ ПОДХОДЫ»
Т.С. Белышева1, Т.В. Наседкина2, И.С. Клецкая3, А.С. Волкова1, В.В. Семенова1, 2, 4, Т.Т. Валиев1
1 Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина, Москва, Российская Федерация
2 Институт молекулярной биологии им. В.А. Энгельгардта РАН, Москва, Российская Федерация
3 Российская детская клиническая больница РНИМУ им. Н.И. Пирогова, Москва, Российская Федерация
4 Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии имени Дмитрия Рогачева Министерства здравоохранения Российской Федерации, Москва, Российская Федерация
Пигментная ксеродерма: клинико-генетические особенности и терапевтические подходы
Белышева Татьяна Сергеевна, доктор медицинских наук, ведущий научный сотрудник научно-консультативного отделения НИИ детской онкологии и гематологии Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина Адрес: 115478, Москва, Каширское ш., д. 24, e-mail: klinderma@bk.ru Статья поступила: 25.09.2021, принята к печати: 17.12.2021
Пигментная ксеродерма — редкое генетическое заболевание, характеризующееся повышенной чувствительностью кожи к повреждающему действию ультрафиолетового (УФ) излучения. У большинства заболевших (до 75%) первые симптомы возникают в раннем возрасте. Характерно хроническое солнечное поражение кожи со стадийностью изменений и риском развития злокачественных опухолей, величина которого зависит от участвующего гена. Помимо изолированных кожных проявлений, описаны неврологические нарушения, включающие прогрессирующие когнитивные дисфункции, нейросенсорную тугоухость, атаксию, пирамидные и экстрапирамидные расстройства, арефлек-сию. Лечение больных с пигментной ксеродермой преимущественно симптоматическое и превентивное (защита от УФ-излучения). В настоящее время ведутся разработки таргетных лекарственных препаратов для репарации ДНК, повышения устойчивости клеток к воздействию УФ-излучения и профилактики таким образом возникновения онкологических заболеваний.
Ключевые слова: пигментная ксеродерма, онкодерматозы, генетические аномалии, дети
Для цитирования: Белышева Т.С., Наседкина Т.В., Клецкая И.С., Волкова А.С., Семенова В.В., Валиев Т.Т. Пигментная ксеродерма: клинико-генетические особенности и терапевтические подходы. Вопросы современной педиатрии. 2021;20(6S):611-617. doi: 10.15690/vsp.v20i6S.2370
Пигментная ксеродерма Xeroderma pigmentosum) — описана в 1874 г. Морицем Капоши [1]. Наиболее часто
генетически опосредованное заболевание с аутосомно- пигментная ксеродерма встречается в Японии (заболева-
рецессивным типом наследования, характеризующееся емость 1 случай на 22 тыс. населения), среди представи-
повышенной чувствительностью кожи к повреждающему телей европеоидной расы болезнь диагностируют гораз-
действию ультрафиолетового (УФ) излучения. Впервые до реже (заболеваемость, по некоторым оценкам, не
Tatyana S. Belysheva1, Tatyana V. Nasedkina2, Iryna S. Kletskaya3, Anastasiya S. Volkova1, Vera V. Semenova1, 2, 4, Timur T. Valiev1
1 National Medical Research Center of Oncology named after N.N. Blokhin, Moscow, Russian Federation
2 Engelhardt Institute of Molecular Biology, Moscow, Russian Federation
3 Russian Children’s Clinical Hospital, Pirogov Russian National Research Medical University, Moscow, Russian Federation
4 Research Center of Pediatric Hematology, Oncology and Immunology named after Dmitry Rogachev, Moscow, Russian Federation
Xeroderma Pigmentosum: Clinical and Genetic Features and Therapeutic Approaches
Xeroderma pigmentosum is rare genetic disorder characterized by increased skin sensitivity to damaging ultraviolet (UV) light. First symptoms manifest at early age in most cases (up to 75%). Chronic damage due to sun exposure is common, it has different stages of changes and risk of further development of malignant tumors that depends on the gene involved. Additionally to skin manifestations there are various neurological disorders such as progressive cognitive dysfunctions, sensorineural hearing loss, ataxia, pyramid and extrapyramidal disorders, areflexia. Treatment of patients with xeroderma pigmentosum is mostly symptomatic and preventive (protection against UV). Nowadays targeted medications for DNA repair and increasing cells resistance to UV light, thus preventing the oncological diseases, are under development.
Keywords: xeroderma pigmentosum, oncodermatosis, genetic anomalies, children
For citation: Belysheva Tatyana S., Nasedkina Tatyana V., Kletskaya Iryna S., Volkova Anastasiya S., Semenova Vera V., Valiev Timur T. Xeroderma Pigmentosum: Clinical and Genetic Features and Therapeutic Approaches. Voprosy sovremennoi pediatrii — Current Pediatrics. 2021;20(6S):611-617. (In Russ). doi: 10.15690/vsp.v20i6S.2370
более 1 случая на 1 млн населения) [2]. Случаи пигментной ксеродермы характерны для географических, национальных и религиозных изолятов и регионов, где практикуют кровнородственные браки [3]. Лица мужского и женского пола болеют одинаково часто [4]. В русскоязычной медицинской литературе опубликованы описания единичных случаев пигментной ксеродермы [5-7], а статистические данные о заболеваемости в России отсутствуют.
Патогенез заболевания впервые был описан Джеймсом Кливером в 1968 г. Он экспериментально доказал способность культуры нормальных фибробластов кожи восстанавливать структуру дезоксирибонуклеино-вой кислоты (ДНК) после повреждающего воздействия УФ-излучения, тогда как фибробласты пациентов с пигментной ксеродермой такой способностью не обладали либо она была значительно снижена [8]. Этот феномен обусловлен недостаточностью ферментов ДНК-эндонуклеазы и ДНК-полимеразы, отвечающих за репарацию фотоповрежденной ДНК [4].
На сегодняшний день известно о восьми комплементарных группах пигментной ксеродермы, выделяемых на основе генетических поломок [4, 9, 10]. Первые 7 комплементарных групп, обозначаемые как XPA-XPG, связаны с нарушением процесса эксцизионной репарации нуклеотидов (исправления ошибочных сшивок нуклео-тидов, возникающих при воздействии УФ-излучения). Различные комплементарные группы отвечают за отдельные этапы процесса репарации: распознавание повреждений — группы XPE (ген DDB2) и XPC (ген XPC); расплетание нитей ДНК — группы XPB (ген ERCC3) и XPD (ген ERCC2); верификация повреждений — группа XPA (ген XPA); расщепление поврежденного участка с различных концов поврежденных оснований — с 5′-кон-ца — группа XPF (ген ERCC4) и с З’-конца — группа XPG (ген ERCC5) [2, 10]. Нарушения эффективности процесса репарации приводят к накоплению изменений ДНК в клетках, что обусловливает высокий риск возникновения онкологических заболеваний — базальноклеточного и плоскоклеточного рака, меланомы [9].
Восьмая комплементарная группа, обозначаемая XPV, ассоциирована с изменениями в гене POLH, кодирующем ДНК-полимеразу, в этих случаях происходит нарушение пострепликационной репарации ДНК [11]. Наиболее часто изменения возникают в генах XPA и XPC, а наибольшая чувствительность к УФ-излучению наблюдается при дефекте гена XPA [11, 12]. В 2020 г. группа китайских исследователей описала изменения еще в одном гене — DDB1, который кодирует большую субъединицу белка DDB (DNA damage-binding protein, или UV-DDB), ответственном за выявление повреждений ДНК в процессе эксцизионной репарации нуклеотидов [13]. Повреждение этого гена приводит к развитию формы пигментной ксеродермы, описанной для подгруппы XPE, для которой в классическом варианте течения характерны изменения в гене DDB2. Данная форма пигментной ксеродермы — одна из наиболее редких [2]. Как правило, она выявляется у взрослых пациентов и отличается высоким риском развития онкологических заболеваний, упомянутых выше [13].
Выделяют два механизма эксцизионной репарации нуклеотидов: репарация всего генома (комплементарные группы A, B, D, F, G) и репарация, связанная с транскрипцией (группы C, E, и V) [3]. Эти механизмы определяют клиническую картину заболевания [1]. В комплементарных группах, патогенетически связанных с нарушением
нуклеотиднои репарации всего генома, чаще отмечаются островыраженные кожные реакции, наблюдаемые с раннего возраста [2]. Соответственно, в этих группах пигментная ксеродерма диагностируется раньше, пациентам раньше начинают проводить фотопротективную терапию, и, как следствие, реже развиваются пигментные изменения кожи и злокачественные новообразования кожи [14]. Напротив, в комплементарных группах, для которых характерно нарушение репарации, связанной с транс-крипциеи, клинические проявления возникают позже [2, 10]. Так, для группы ХРС характерны лентигинозные изменения кожи, возникающие ближе к двухлетнему возрасту [15]. Накопление фотоповреждений вследствие установления диагноза в относительно позднем возрасте ассоциировано с высоким риском онкологических заболеваний кожи [4]. В группах ХРЕ и ХРУ клинические проявления болезни могут возникать ближе ко второму десятку лет жизни, что также приводит к большему накоплению изменений в генах. Таким образом, несмотря на более благоприятное течение заболевания в комплементарных группах ХРС, ХРЕ, ХРУ, у таких больных опухоли кожи в последующем развиваются чаще.
Выделяют следующие клинические варианты пигментной ксеродермы: классический с кожными проявлениями, пигментная ксеродерма с неврологическими симптомами (нейросенсорная глухота, психические нарушения, атаксия), пигментная ксеродерма с симптомами трихотиоди-строфии, пигментная ксеродерма с синдромом Кокейна и цереброокулофациоскелетный синдром [16, 17]. У 75% пациентов заболевание манифестирует в возрасте от 6 мес до 3 лет, у 5% — в возрасте старше 14 лет [18].
Острые кожные реакции возникают не у всех пациентов и чаще — у младенцев в виде стойкой эритемы, отечности с формированием везикул — признаков солнечных ожогов кожи. Часто в раннем детском возрасте обнаруживается светобоязнь. Родители больного ребенка отмечают повышенную восприимчивость к свету, слезотечение, непроизвольные почесывания век, отек и гиперемию конъюнктивы глазных яблок, раздражительность и плаксивость. Дети предпочитают затемненное место в комнате, избегают яркого солнечного света. В дальнейшем в процесс вовлекается роговица глаза, формируются очаги хронического воспаления на конъюнктиве, веках, развиваются злокачественные новообразования. Многократные эпизоды хирургического лечения по поводу прогрессирования опухолевого процесса приводят к развитию рубцовых изменений. Может происходить сращение конъюнктивального мешка и укорочение конъ-юнктивальных сводов, часто наблюдается сращение краев век, в дальнейшем может развиться офтальмоскле-роз. Все эти изменения приводят к снижению и даже потере зрения.
Хроническое солнечное поражение кожи развивается у всех больных с пигментной ксеродермой и имеет характерную стадийность. Обычно у детей в возрасте двух и более лет уже можно наблюдать пигментные высыпания по типу эфелидов (веснушек) или лентиго темно- и светло-коричневого цвета, пятна диаметром до 3-5 мм на открытых участках кожи. В дальнейшем в этих зонах формируется картина атрофических изменений — истончение, сухость кожи, множественные телеангиэкта-зии, очаги гипо- и гиперпигментации (пойкилодермия), кожа становится более грубой, приобретает пестрый вид, теряет способность собираться в складку. В области головы отмечается изменение подлежащей хрящевой
ткани — деформация и истончение носа, ушных раковин, уменьшаются размеры носовых отверстий. Развивается микростомия (уменьшение размеров рта), актинический хейлит — стойкое воспаление красной каймы губ, ее истончение. Позже формируются гиперкератотические и веррукозные (бородавчатые) разрастания, которые расцениваются как предраковые. Финализирует процесс стадия злокачественных опухолей. Отмечено, что у пациентов с остро протекающими кожными реакциями и наличием неврологической симптоматики реже развивается рак кожи в связи с ранней диагностикой и инициацией профилактических мероприятий [15].
Останавливаясь на возможных сочетаниях пигментной ксеродермы и нейродегенеративных изменений, можно отметить наиболее частые виды поражений нервной системы: прогрессирующие когнитивные нарушения, нейросенсорную тугоухость, атаксию, пирамидные и экстрапирамидные расстройства, арефлексию. Известно, что в Японии сочетание кожных проявлений и неврологической симптоматики имеется у 55% больных, тогда как у пациентов из западных стран — в 25% случаев [19]. Чаще всего неврологическая симптоматика присутствует в комплементарных группах ХРА и XPD [9-11].
Описано также сочетание пигментной ксеродермы и синдрома Кокейна [20, 21]. Как самостоятельная нозологическая единица синдром Кокейна — это редкое мультисистемное дегенеративное заболевание. Спектр клинических проявлений синдрома обширен и включает светочувствительность, нейродегенератив-ные изменения, контрактуры суставов, нарушения слуха. Патогенетическая основа синдрома Кокейна — изменения структуры генов ERCC6 и ERCC8, приводящие к нарушению репарации ДНК [19]. При этом, в отличие от пигментной ксеродермы, у пациентов с синдромом Кокейна гораздо реже развиваются злокачественные опухоли [22]. Сочетание пигментной ксеродермы и синдрома Кокейна (ХР/СБ-комплекс) имеет ряд особенностей: развитие прогерии, наличие неврологических нарушений в связи с течением тигроидной демиелинизации — одной из форм лейкодистрофии. Важно отметить, что в случае ХР/СБ-комплекса отсутствуют изменения в структуре гена, приводящего к развитию синдрома Кокейна, т.е. ХР/СБ-комплекс — это сочетание фенотипических проявлений двух заболеваний с генотипом, характерным для пигментной ксеродермы (описаны изменения в комплементарных группах ХРВ, ХРО, XPF и XPG) [20]. ХР/СБ-ком-плекс развивается крайне редко. В период с 1965 по 2021 г. описано 44 таких клинических случая [20-22].
Ранее уже указывалось, что при пигментной ксе-родерме значительно возрастает риск возникновения злокачественных новообразований, в том числе базаль-ноклеточной карциномы, меланомы и плоскоклеточного рака кожи. Описано несколько случаев пигментной ксе-родермы, ассоциированной с ангиосаркомой [23-25]. Учитывая, что воздействие УФ-излучения не является фактором риска возникновения ангиосаркомы [24], связь между этими двумя заболеваниями остается неясной. Приводятся случаи возникновения ангиосаркомы на поврежденных участках кожи, а также был описан один случай этого заболевания с поражением языка [11, 25]. В качестве примера развития злокачественных новообразований кожи рассмотрим клинический случай пигментной ксеродермы у ребенка 8 лет.
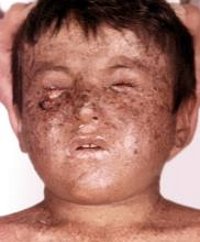
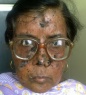
Пациент Б., 8 лет, гражданин Кыргызстана, обратился в федеральный клинический центр в августе 2014 г.
с диагнозом «пигментная ксеродерма». При осмотре на коже лица, шеи, волосистой части головы, плечах, верхних и нижних конечностях определяются множественные темно- и светло-коричневые пятна размером 3-5 мм. В области лица и шеи на коже участки атрофии и гиперкератоза. На голове волосы выраженно разрежены, местами полностью отсутствуют. Лицо отечно, глаза не открываются из-за синехий. На правой щеке глубокая язва размером 3 х 2 х 2 см с небольшим количеством гнойного отделяемого, переходящая на правую височную область. В правой периорбитальной области опухолевое образование багрового цвета с эрозиро-ванной поверхностью размером 3 х 1 х 1 см. На коже лба справа и в левой височной области опухолевые разрастания с неровной поверхностью, диаметром около 2 см. В области красной каймы нижней губы справа визуализируется папилломатозное опухолевое образование, диаметром 1 см, покрытое желтоватой корочкой, слева — язва с некротическими очагами. Правая ушная раковина выраженно деформирована, на внутренней поверхности определяются язвенные дефекты. Нос также значительно деформирован, на коже визуализируется язва, покрытая плотной коркой. Пациент предъявляет жалобы на болевой синдром, частичную потерю слуха, наличие опухолей в области лица. Из анамнеза: ребенок от второй беременности, вторых самостоятельных родов. Масса тела при рождении — 2900 г, рост — 50 см. Раннее развитие без особенностей. Семейный анамнез не отягощен. В возрасте 6 мес родители заметили у ребенка светобоязнь, с полутора лет начали появляться участки гиперпигментации по типу веснушек на открытых участках кожи. В возрасте 4 лет родители пациента впервые обратились в медицинское учреждение по месту жительства по поводу опухолевого образования на коже века в области правого глаза ребенка. Проведено хирургическое лечение, по результатам гистологического исследования — плоскоклеточный рак кожи. Вскоре после первого эпизода на коже лица начали появляться новые опухолевые образования. Клинический диагноз: «пигментная ксеродерма». В 2011-2013 гг. ребе-
Рисунок. Пациент Б., 8 лет, с пигментной ксеродермой Figure. Patient B., 8 years old, with xeroderma pigmentosum
Предрак кожи — пигментная ксеродерма

Пигментная ксеродерма — тяжелое генетическое заболевание кожи. Оно возникает у маленьких детей — как только они начинают активно “знакомиться” с ультрафиолетовым излучением.
Поговорим, как проявляется пигментная ксеродерма, как она лечится и какой у нее прогноз.
Причины заболевания
Пигментная ксеродерма — заболевание, поражающее детей кровнородственных браков. В этом случае у ребенка в коже отсутствует защита от ультрафиолетовых лучей.
Когда кожа здорового человека сталкивается с ультрафиолетом, в ДНК возникает повреждение. Оно распознается, приходит специальный фермент и вырезает его. Дальше включается другой фермент. Он воспроизводит тот участок, который был до повреждения, а третий фермент “пришивает” вновь образованный кусочек в “разорванное” место.
При пигментной ксеродерме этих процессов не происходит. В ДНК клеток раз за разом возникают поврежденные участки. И, когда в ключевых генах накапливается 5-10 повреждений, клетка кожи подвергается злокачественному перерождению.
Пигментная ксеродерма чаще всего перерождается в плоскоклеточный рак. Реже она трансформируется в меланому, ангиосаркому, саркому, трихоэпителиому или базально-клеточный рак. Иногда онкодерматологи докладывают о том, что у отдельных людей встречаются сразу несколько типов рака кожи.
Как передается заболевание
Есть два типа пигментной ксеродермы. Одна передается аутосомно-доминантным путем, то есть, если ген есть только у одного из родителей, то у ребенка шанс заболеть — от 50 до 100%. Вторая — аутосомно-рецессивным путем. Здесь шанс заболеть — от 0 до 25%.
В конце прошлого года я обнаружила, что малейшие ранки на моих руках сразу воспаляются и гноятся. Незамедлительно обратившись к специалисту, я по его рецепту начала пользоваться мазью.
Симптомы заболевания
Почти ¾ всех случаев ксеродермы зафиксировано у детей в возрасте 6-12 месяцев. Первые симптомы появляются весной или летом, когда с ребенком начинают активно гулять,открывая его кожу ультрафиолетовым лучам.
Пик заболеваемости приходится на 8-летний возраст. Зафиксированы редкие случаи, когда болезнь развивалась у подростка, во взрослом возрасте или даже на 65-м году жизни.
Пигментная ксеродерма развивается таким образом:
❶ Сначала на тех участках кожи, куда попал солнечный свет, появляется покраснение и отек. Вскоре здесь возникают пузыри различной величины.
❷ Когда пузыри и отек исчезает, кожа не светлеет. На ней остаются пятна желтого, коричневого или бурого цвета. Они напоминают веснушки.
❸ Если пораженная кожа снова получает дозу ультрафиолетового излучения, вновь возникают отек, покраснение и пузыри. После этого кожа начинает истончаться. Она сухая, натянута, не собирается в складки. На ней возникают трещины и рубцы.
Если повреждается кожа лица, то стягивается кожа вокруг рта и ноздрей. Кончик носа и мочки ушей становятся тоньше. Возникают конъюнктивиты, кератиты, блефариты. На коже век могут появляться сосудистые звездочки.
❹ Далее на истонченной коже появляются новообразования: бородавки, папилломы, кератомы, фибромы.
❺ Через 10-15 лет, а иногда и раньше на фоне ксеродермы развиваются злокачественные опухоли. Они быстро метастазируют во внутренние органы. От этого наступает смерть.
Лечение
К сожалению, методов лечения не разработано. При первых же симптомах заболевания человека надо защитить от ультрафиолетовых лучей:
► обрабатывать кожу мазями, которые содержат или хинин, или салол, или парааминобензойную кислоту. Периодически вместо них использовать солнцезащитные крема с SPF 40-50;
► при выходе на улицу надевать только закрытую одежду из плотной ткани, широкополые шляпы, перчатки, солнцезащитные очки;
► покрыть стекла в автомобиле и квартире больного такой пленкой, которая не пропустит ультрафиолетовых лучей;
► запретить ему выходить утром и днем;
► не использовать галогеновые и энергосберегающие лампы.
Как только появятся бородавки или прочие новообразования, их нужно удалять. При развитии злокачественных опухолей — их нужно срочно лечить.
Поскольку больной вынужден жить в темноте, ему нужно принимать витамин D в соответствующей дозировке.
Прогноз
Заболевание имеет прогрессирующее течение. Оно может приостанавливаться, могут возникать периоды улучшения. Около 66% детей с пигментной кератомой умирают от метастазов в возрасте до 15 лет.
Тем не менее, примерно 45% людей с пигментной кератомой все же доживают до 40 лет. Некоторые живут до старости, если им удается сберечь кожу от солнца.
Пигментная ксеродерма
Пигментная ксеродерма (лентикулярный меланоз, злокачественное лентиго, пигментная атрофодермия) — хроническое наследственное заболевание, обусловленное повышенной чувствительностью кожи к солнечной радиации и УФ-лучам. Изменения кожи характеризуются последовательно сменяющими друг друга процессами воспаления, гиперпигментации, атрофии, гиперкератоза и злокачественной трансформации клеток кожи. У большинства больных отмечается поражение глаз: конъюнктивит, кератит и опухоли. Диагноз устанавливается при выявлении повышенной чувствительности кожи к УФ и характерной гистологической картины. Лечение симптоматическое.
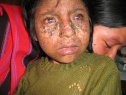

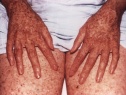
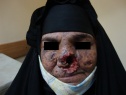
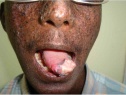
Общие сведения
По данным статистических исследований, которые проводит дерматология, пигментная ксеродерма встречается в среднем с частотой 1 случай на 250 тыс. человек. Наиболее высокая заболеваемость отмечается среди населения стран Среднего Востока и Средиземноморского побережья Африки. Пигментной ксеродермой заболевают оба пола, однако некоторые дерматологи отмечают, что среди девочек болезнь встречается чаще. Заболевание часто носит семейный характер и наблюдается при близкородственных браках.
Причины возникновения пигментной ксеродермы
Различают 2 генотипа пигментной ксеродермы: передающийся аутосомно-доминантным путем и аутосомно-рецессивным. В основе заболевания лежит генетически обусловленная недостаточность ферментов УФ-эндонуклеазы и полимеразы-1, которые отвечают за восстановление ДНК после ее повреждения УФ-лучами. В связи с этим у больных после пребывания в условиях повышенной солнечной радиации развиваются поражения кожи и глаз. У некоторых гетерозиготных носителей гена наблюдаются стертые формы пигментной ксеродермы в виде различных пигментных нарушений.
Симптомы пигментной ксеродермы
Наиболее часто (в 75% случаев) начало пигментной ксеродермы приходится на первые 6-12 месяцев жизни. Заболевание проявляется весной или летом, как только ребенок попадает под интенсивное воздействие солнечных лучей. В отдельных случаях начальные проявления пигментной ксеродермы наблюдались у пациентов в возрасте 14-35 лет и даже на 65-ом году жизни.
В течении пигментной ксеродермы выделяют 5 переходящих одна в другую стадий: зритематозную, стадию гиперпигментаций, атрофическую, гиперкератическую и стадию злокачественных опухолей.
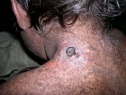
Эритематозная стадия характеризуется воспалительными изменениями кожи на участках, подвергшихся воздействию ультрафиолета. В таких областях кожного покрова появляются покраснение, отек, мелкие пузырьки и пузыри. После разрешения элементов первой стадии на коже остаются коричневые, желтоватые или бурые пигментные пятна, похожие на веснушки (стадия гиперпигментаций).
Последующие облучения кожи у пациентов с пигментной ксеродермой ведут к новым воспалительным и пигментным изменениям кожи, что вызывает развитие атрофических процессов. Заболевание переходит в атрофическую стадию, для которой характерны истончение и сухость кожи, образование трещин и рубцов. Кожа натянута и не собирается в складки. Отмечается уменьшение рта (микротомия), атрезия отверстия рта и носа, истончение кончика носа и ушей.
Гиперкератическая стадия проявляется развитием в очагах пораженной кожи бородавчатых разрастаний, папиллом, кератом, фибром. Стадия злокачественных опухолей наблюдается, как правило, спустя 10-15 лет от начала пигментной ксеродермы. Но иногда злокачественные новообразования появляются в первые годы заболевания. К злокачественным опухолям, встречающимся при пигментной ксеродерме, относятся базалиомы, меланомы, эндотелиомы, саркомы, трихоэпителиомы, ангиосаркомы. Они характеризуются быстрым метастазированием во внутренние органы, приводящим заболевание к летальному исходу.
В 80-85% случаев пигментной ксеродермы наблюдаются поражения глаз в виде конъюнктивита, кератита, гиперпигментации и атрофии радужки и роговицы, приводящих к понижению зрения. На коже век появляются телеангиэктазии, гиперкератозы, дисхромии и опухолевые процессы. Нередко пигментная ксеродерма сочетается с дистрофическими изменениями тканей: дистрофией зубов, синдактилией, врожденной алопецией, отставанием в росте. Пигментная ксеродерма, сопровождающаяся умственной отсталостью ребенка, выделена как отдельная клиническая форма — синдром де Санктиса-Какионе.
Диагностика пигментной ксеродермы
Специфический метод диагностики пигментной ксеродермы проводится при помощи монохроматора и заключается в выявлении повышенной чувствительности кожного покрова к воздействию ультрафиолета.
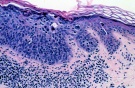
Для уточнения диагноза дерматолог назначает биопсию пораженного участка кожи. Последующее гистологическое исследование в ранней стадии заболевания определяет гиперкератоз, отек и воспалительную инфильтрацию дермы, истончение росткового слоя, пигментацию базального слоя. В атрофической и гиперкератической стадии наблюдается атрофия эпидермиса, дегенеративные изменения коллагеновых и эластических волокон. В стадии злокачественных опухолей — атипические клетки и гистологическая картина рака кожи.
Лечение пигментной ксеродермы
Пациентам следует избегать воздействия УФ-лучей: носить шляпы с большими полями и вуали, применять солнцезащитные крема и мази, использовать пудры с танином. Медикаментозное лечение пигментной ксеродермы в основном симптоматическое и, к сожалению, малоэффективно. Применяют ароматические ретиноиды, токоферол, хингамин. При развитии злокачественных процессов дополнительно назначают проспидин, пиридоксин, тиамин, цианокобаламин. Папилломатозные и бородавчатые разрастания удаляют хирургически, путем криодеструкции, электрокоагуляции или удаления лазером.





